
研究背景:
近年来,铅基卤化物钙钛矿APbX3(A = CH3NH3, (H2N)2CH, Cs; X = I, Br, Cl)在光伏、发光及探测等领域展现出优异的性能。其中[PbX6]金属卤素八面体被视为功能单元,Pb2+独特的价层电子构型和[PbX6]八面体三维顶角连接特性是这类物质具有优异性能的重要原因。虽然铅基钙钛矿性能优异但是面临着稳定性较差和不环保的劣势,这促使研究者们探索其他稳定且高效的卤化物钙钛矿及其衍生物。其中,卤化物双钙钛矿A2B(I)B(III)X6维持了单钙钛矿三维的晶体结构,并具有稳定性好且不含铅元素的特点,在光致发光及X-ray探测等领域展现出优异的性能。由于B位阳离子对卤化物双钙钛矿的性能起着决定性的作用,以往工作基于B(I)和B(III)元素组合的策略对卤化物双钙钛矿新材料进行了大量的拓展,而性能调控则是通过对B位置阳离子的掺杂及固溶。但是从电子维度的角度,A2B(I)B(III)X6型双钙钛矿的电子维度是零维或者准零维,因为B(I)和B(III)位阳离子的前线轨道之间要么存在能量不匹配问题(比如Cs2NaSbCl6)要么存在对称性不匹配问题(比如Cs2AgBiCl6),这使得贡献价带顶和导带底原子轨道的连接性较差。因而相对APbX3型单钙钛矿来说,A2B(I)B(III)X6型双钙钛矿材料的能带带边相对局域化、光学带隙大、载流子有效质量大以及电荷的传输与分离相对困难。 因此,除了元素组合策略之外,进一步改变B位离子的排列方式来拓展双钙钛矿的结构维度并进一步调控电子结构是一个重要但是鲜有关注的研究方向。
成果简介:
近日,肖泽文教授课题组创新性地提出了一种调控卤化物双钙钛矿A2B(I)B(III)X6的B位阳离子排列方式的策略来改善其电子维度进而调控其电子性质。作者从岩盐矿构型(零维)出发,对B位置阳离子重排构建了柱状(一维)和层状(二维)排列的双钙钛矿结构模型(图a),并对它们的热力学稳定性和电子性质进行理论计算评估。DFT计算表明调控B位阳离子的排列方式可以有效地调节电子结构(图b),但是热力学稳定性问题是制约这类材料的合成,比如A2B(I)B(III)X6双钙钛矿采用柱状和层状排列构型时热力学稳定性逐步降低(图a)。通过对稳定性相对较优的柱状构型进行结构分析后,作者首次提出阴离子空位有序的B位阳离子柱状排列的双钙钛矿A2B(I)B(II)X5模型(图c),理论计算表明降低电荷差异提升了这种模型的热力学稳定性。在初步选定各个位置的元素后,利用DFT计算预测出这些材料的热力学稳定性顺序并筛选出几个可能稳定的化合物(图d)。实验成功合成出Cs2AgPdCl5、Cs2AgPdBr5和Cs2AgPtCl5三种新型柱状双钙钛矿材料,这验证了理论计算的合理性,实现了理论设计与实验验证。这些材料具有较低的光学带隙(1.33-1.77 eV),可以有效地吸收可见光,同时沿着同种八面体链的方向有效质量较小。此外,这些双钙钛矿材料表现出优异的热稳定性和空气稳定性。该工作是理论与实验结合的模范案例,提出的新型柱状卤化物双钙钛矿结构原型有望作为一个新的平台来探索更多的钙钛矿半导体光电材料。
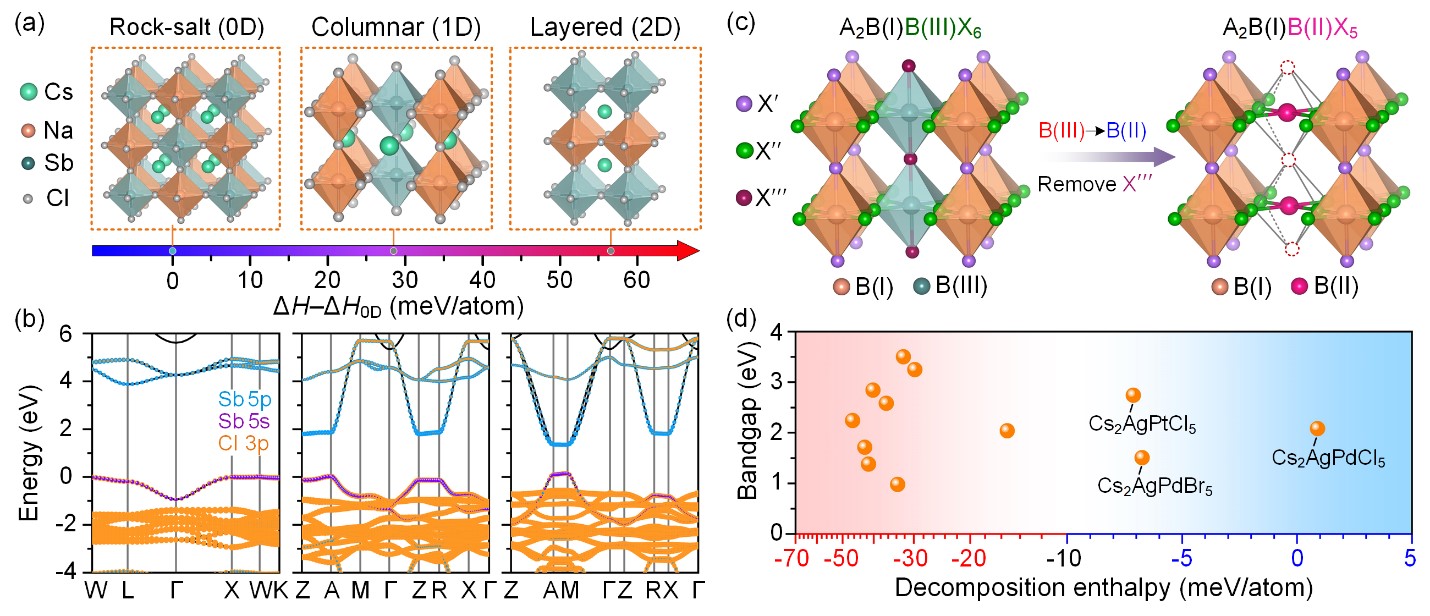
图1.不同构型Cs2NaSbCl6的(a)分解焓和(b)能带结构,(c)从B位柱状排列的A2B(I)B(III)X6型双钙钛矿到A2B(I)B(II)X5型双钙钛矿的结构演变,(d)设计的B位柱状排列的双钙钛矿Cs2B(I)B(II)X5(B(I)= Ag, Na; B(II)=Pd, Pt; X=Cl, Br, I)的带隙和分解焓。
该工作第一完成单位为华中科技大学武汉光电国家研究中心,肖泽文教授为论文唯一通讯作者,其博士生吉国奇为第一作者。
论文信息:
B-Site Columnar-Ordered Halide Double Perovskites: Theoretical Design and Experimental Verification,DOI:10.1021/jacs.1c03825. 论文链接:https://pubs.acs.org/page/pdf_proof?ref=pdf




